
Medical Biochemistry
Research
Through metabolism, biochemical processes give rise to the complexity of life. Acquired and inborn errors in metabolism underlie many diseases occurring in man. The challenge for present day medical biochemistry is to find, and integrate, pieces of information at molecular, cell and organismal level to increase understanding of biochemical processes in health and disease. Ultimate proof of insight is discovery of applicable biomarkers and rational design of effective therapeutic interventions for metabolic disorders. We aim to further accomplish these by multidisciplinary research in (inter)national collaboration with experts in different fields. In recent years, chemical biology and analytical chemistry have increasingly expanded our research tools. Our current work focuses on glycosphingolipids and their metabolizing enzymes, a topic relevant for lysosomal storage disorders, neurodegenerative disease and metabolic syndrome.
Unique metabolites in the human body are glycosphingolipids, membrane lipids largely residing at the cell surface with their sugars facing the external milieu. Glycosphingolipids together with cholesterol form transient semi-ordered microdomains in which specific proteins, including signaling receptors, preferentially reside. In all cells, glycosphingolipids are continuously synthesized at the ER and Golgi apparatus and degraded inside lysosomes.
The role of glycosphingolipids in health and disease is our central research theme. Inherited defects in lysosomal degradation of glycosphingolipids cause sphingolipidoses, a number of discrete lysosomal storage disorders in man. Highlight of our research are studies on Gaucher disease, a relatively common sphingolipidosis due to impaired glucocerebrosidase activity. Our longstanding investigation of glucocerebrosidase and Gaucher disease has a multidisciplinary and translational nature, involving collaborations with academic colleagues from different disciplines as well as patient societies and industry. Topics of investigation range from lipid substrates and products of glucocerebrosidase, to regulation of synthesis, structural features and life cycle of (mutant) protein in various cells, to mechanisms translating enzymatic deficiency into neurological, epidermal, skeletal, bone marrow, splenic and hepatic symptoms of Gaucher patients. In addition, considerable attention has been paid to discovery of now widely applied biomarkers for Gaucher disease and development of now registered therapies. We still pursue these research lines to further depth and extend them to other sphingolipidoses like Fabry disease and Krabbe disease using various research models like patient materials, mice, zebra fish, and cultured cells.
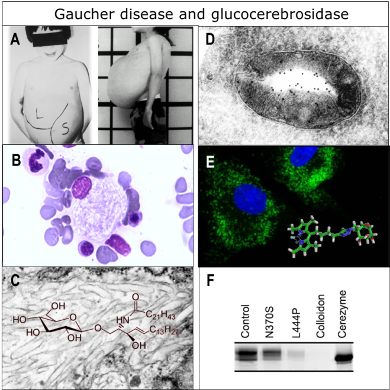
Fig. 1: Gaucher disease
A. Patient suffering from type 1 Gaucher disease showing characteristic hepatosplenomegaly.
B. Typical pathological storage macrophage in GD patients (Gaucher cell).
C. Electron micrograph showing tubular glucosylceramide deposits in GD spleen.
D. ImmunoEM localization of glucocerebrosidase (GBA) in lysosome of human macrophage.
E. In vivo ABP-labeling of active GBA molecules in lysosomes of cultured fibroblasts. ABP shown as insert.
F. Comparison of active GBA in fibroblast of normal subject patients with mild, severe and acutely lethal disease. Cerezyme: therapeutic recombinant GBA.
Defects in lysosomal glucocerebrosidase, even at the level of carriers, have been recently recognized as an important genetic risk factor for relatively common alpha-synucleinopathies such as Parkinson disease and Lewy-body dementia. Moreover, it has become clear that acquired deregulation of glycosphingolipid metabolism occurs in obese individuals and is linked to manifestations of the metabolic syndrome, a multifactorial pathology with insulin resistance, hepatosteatosis and atherosclerosis as prominent symptoms. Abnormally elevated glycosphingolipids during obesity appear to affect the normal regulation of body metabolism and satiety, propagating the unhealthy state. Pharmacological intervention in glycosphingolipid metabolism was shown by us to offer protection in obese rodents against various manifestations of the metabolic syndrome. In collaboration with Prof. Herman Overkleeft (BIOSYN/LIC), iminosugar-type modulators of key enzymes in glycosphingolipid metabolism are further improved as potential therapeutic agents for lysosomal storage disorders, neurodegenerative alpha-synucleinopathies and metabolic syndrome manifestations.
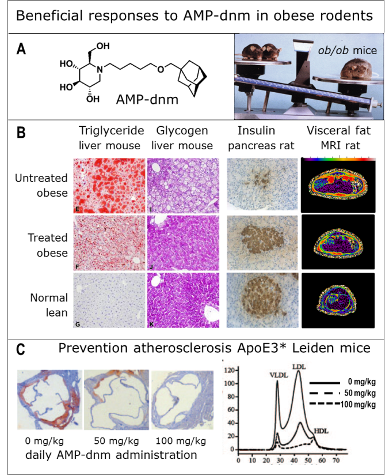
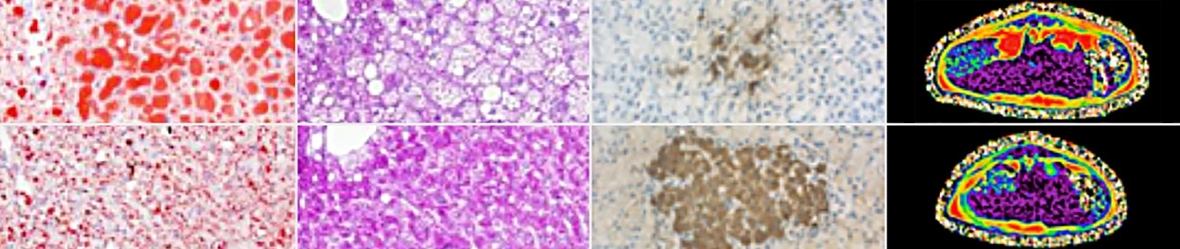
Fig. 2: Aza-sugar treatment of metabolic syndrome
A. Structure of therapeutic azasugar AMP-DNM (left); obese (ob/ob) mice suffering from leptin-deficiency used as research model.
B. Examples of beneficial responses to oral AMP-dnm treatment in obese ob/ob mice and ZDF rats (left to right): Reduction of excessive liver triglyceride and increase of reduced liver glycogen in ob/ob mice, protection of insulin-producing beta-cells in pancreasand reduction of harmful visceral fat in ZDF rats.
C. Protection against atherosclerosis by oral AMP-dnm treatment in ApoE3*Leiden mice on high fat diet (left). Drug-induced reduction in harmful LDL cholesterol (right).
In our fundamental and translational investigation of glycosidases, we increasingly employ smart chemical biology tools designed with Prof. Herman Overkleeft. Briefly, fluorescent activity-based probes allow us to monitor in vivo active glycosidase molecules with unprecedented sensitivity and spatial detail. Various applications for these tools are found, increasing our insight in real-time properties of enzymes in their true environment in cells of living organisms. Our notion that in vivo some glycosidases also act as transglycosylase, generating hitherto unknown glycosylated metabolites, opens new research avenues on (patho)physiological functions of such novel compounds. Superior in situ imaging of active glycosidase molecules and various related lipid metabolites in living organisms, combined with pharmacological manipulation of these biochemical processes, is the prime focus of our research group.