
Going accurate for molecule – metal surface interactions
Researchers from the THEOR CHEM group at Leiden University strive to set new benchmarks in the accuracy of the prediction of interaction energies between molecules and metal surfaces.
To predict the efficiency of a catalytic reaction on a computer instead of performing thousands of trial-and-error experiments — this is half dream and half reality at THEOR CHEM, where researchers investigate the driving forces behind catalytic reactions by means of computer simulations. For quantitatively accurate prediction, however, a main ingredient is accurate values of the molecule-metal surface interaction. And these are hard to get.
The interaction between molecules and metal surfaces is governed by quantum mechanical effects. These are captured by the electronic Schrödinger equation. Unfortunately, for large systems, this equation is too difficult to solve, even with the most modern computers in the world. Density functional theory (DFT), a well known and computationally cheap approximation to the electronic Schrödinger is often not accurate enough for the problems considered at THEOR CHEM and the big question is: Can one do better than DFT?
The researchers from THEOR CHEM have taken up the challenge: By applying so-called Quantum Monte Carlo methods (QMC), they approach the problem in a stochastic way and gauge the attainable accuracy.
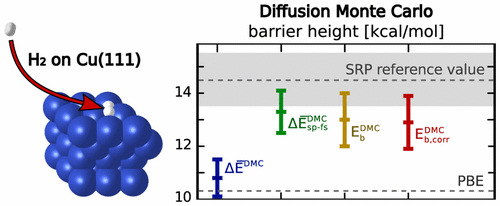
Their recent research, published in the Journal of Computational and Theoretical Chemistry, clearly shows the potential of QMC problems in heterogeneous catalysis. For the system investigated (H2 on Cu(111) ), they obtained an accuracy of ~1.5kcal/mol, close to the accuracy they are ultimately striving for. The fly in the ointment: On an average PC, these calculations would take around 6 million hours.
Nevertheless, this result is an important result on the way towards chemically accurate computations of molecules - metal surface interactions, and it is a first step towards fully ab-initio predictions of heterogeneous catalysis.