
Research project
Modeling energy conversion dynamics at interfaces
Chemical reactions go hand-in-hand with an energy exchange with the environment in which they take place. Surfaces offer a variety of energy dissipation channels, constituted by the nuclear and electronic degrees of freedom of the atoms at the interface. Aiming at an improved future harvesting of energy, Joerg Meyer develops and applies computational methods to obtain a better fundamental understanding of interfacial energy conversion dynamics.
- Contact
- Jörg Meyer
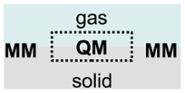
Quantum mechanical (QM) modeling is required in order to accurately account for bond breaking and making during chemical reactions. Density-functional theory (DFT) in particular has proven to be a very successful tool in that respect, in particular when it comes to the description of metallic surfaces. Still, its computational effort limits the time and length scale that can be modeled in dynamical simulations. Already a simple back-of-the-envelope calculation using the speed of sound allows to estimate that these limitations can affect the description of heat transport in the solid via vibrations of the lattice (phonons). With our recently developed new “QM/Me” embedding scheme [2], which links QM- modeling to molecular mechanics (MM) for metallic systems, we can overcome such limitations.
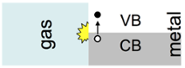
Since there is no energetic gap between the valence band (VB) and the conduction band (CB) in metallic systems, chemical dynamics at the interface can easily excite electrons. This leads to the violation of one of the most fundamental approximations in theoretical modeling, the Born-Oppenheimer approximation, and provides another elementary channel for energy dissipation. Characterizing and quantifying these electron- hole-pair excitations [4] and including their effect on dynamics at an interface [1] poses a formidable theoretical challenge, which we are facing by the development and application of new computational methods.
Key publications
- Rittmeyer, S.P., J. Meyer, I.J. Juaristi, K. Reuter, "Electronic Friction-Based Vibrational Lifetimes of Molecular Adsorbates: Beyond the Independent-Atom Approximation", Physical Review Letters, vol. 115, issue 4, 7/2015. DOI: 10.1103/PhysRevLett.115.046102
- Meyer, J., K. Reuter, "Modellierung von Wärmedissipation auf der Nanoskala: ein Einbettungsansatz für chemische Reaktionen auf Metalloberflächen", Angewandte Chemie, vol. 126, issue 18, pp. 4813 - 4816, 04/2014. DOI: 10.1002/ange.201400066
- Carbogno, C., A. Gross, J. Meyer, K. Reuter, "Adsorption Dynamics at Metal Surfaces: Non-Adiabatic Effects, Dissociation and Dissipation", Dynamics of Gas-Surface Interactions: Atomic-level Description of Elementary Processes, Berlin, Springer, 2012.
- Meyer, J., K. Reuter, "Electron–hole pairs during the adsorption dynamics of O2 on Pd(100): exciting or not?", New Journal of Physics, vol. 13, issue 8, pp. 085010, 08/2011. DOI: 10.1088/1367-2630/13/8/085010
- McNellis, E.R., J. Meyer, K. Reuter, "Azobenzene at coinage metal surfaces: Role of dispersive van der Waals interactions", Physical Review B, vol. 80, issue 20, 11/2009. DOI: 10.1103/PhysRevB.80.205414