
Research project
Dissociative chemisorption on transition metal surfaces
The dissociative chemisorption of a molecule on a transition metal surface represents a rate-limiting step in many heterogeneously catalyzed processes, whereby most chemicals are made. In spite of the importance of this reaction, an accurate first principles approach to modeling it does not yet exist. Challenges that theorists face are that energy transfer to surface vibrations is usually important for any reacting molecule heavier than H2, and that electronically non-adiabatic processes like electron-hole pair excitation may also have to be modeled.
- Contact
- Geert-Jan Kroes
Furthermore, accurately treating all degrees of freedom of the incident molecule with quantum dynamics has thus far only been achieved for diatomic and triatomic molecules. Last but not least, computing potential energy surfaces or forces for direct dynamics simulations is most efficiently done with semi-local density functional theory, but the standard functionals at these levels do not deliver chemical accuracy (1 kcal/mol) for adsorption energies and reaction barrier heights.
We tackle the last problem described with a semi-empirical version of specific reaction parameter density functional theory (SRP-DFT). Here, the focus is on the accurate determination of reaction barrier heights. The SRP-DFT method has already allowed the description of several dissociative chemisorption reactions with chemical accuracy. Demonstrating this requires the use of not only SRP-DFT, but also of dynamics methods, as reaction barriers heights do not represent observables. Instead, demonstrating the accuracy of these quantities requires performing a simulation of a supersonic molecular beam experiment, and showing that the measured dissociative chemisorption (or "sticking") probability as a function of incidence energy curve is reproduced to within an energy shift of less than 1 kcal/mol.
Depending on the requirements of the system, we work with different dynamical models, which may feature energy transfer involving surface atom vibrations and/or electron-hole pairs. To model energy dissipation, on many occasions we collaborate with group member Jörg Meyer. We use both quantum dynamics (often in collaboration with group member Mark Somers) and quasi-classical dynamics methods. When modeling surface atom vibrations, in some cases we use direct dynamics methods like density functional molecular dynamics (DFMD), while in other cases we use high-dimensional neural network (HDNN) potentials and perform quasi-classical trajectory calculations. When also modeling electron-hole pair excitation, we typically use an electronic friction (EF) model, and we have used both DFMD with electronic friction (DFMDEF) and molecular dynamics with electronic friction (MDEF) in combination with HDNN potentials. Advancing to more accurate electronic structure methods, we have also turned to the use of Quantum Monte-Carlo, in collaboration with group member Katharina Doblhoff-Dier.
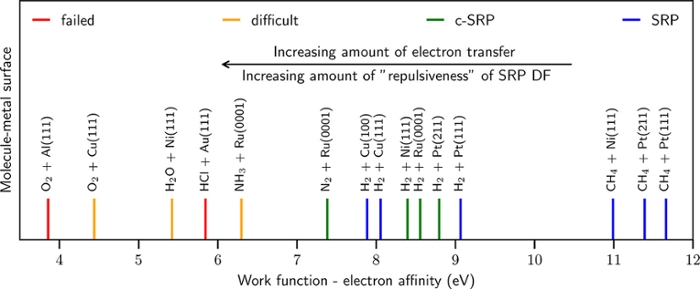
So far we have been quite successful at describing dissociative chemisorption reactions of H2, N2, and CH4 with transition metal surfaces. In all of these systems, the relationship (WF-EA) > 7 eV was obeyed, where WF is the work function of the metal, and EA the electron affinity of the molecule (see the Figure, and N. Gerrits, E.W.F. Smeets, S. Vuckovic, A.D. Powell, K. Doblhoff-Dier, and G.J. Kroes, "DFT for molecule-metal surface reactions: When does the GGA get it right, and what to do if it doesn't", J. Phys. Chem. Lett. 2020, 11, 10552-10560). We are now turning to systems with (WF-EA) < 7 eV. These systems are prone to electron transfer, and therefore to electronically non-adiabatic effects like electron-hole pair excitation. Additionally, these systems are hard, if not impossible, to describe with the standard (GGA and meta-GGA) density functionals of DFT. Overall ideas on how progress with this class of reactions should be achieved (which includes the dissociative chemisorption of many oxygen containing molecules, including reactions that could be important to achieving a sustainable society) are described on Kroes' personal website.
A description of the progress achieved in recent years has been provided in a review paper (G.J. Kroes, " Computational approaches to dissociative chemisorption on metals: Towards chemical accuracy", Phys. Chem. Chem. Phys. 23, 8962-9048, 2021).