Centre for Computational Life Sciences (CCLS)
Flagships
In CCLS several subgroups have formed, below you can find an overview of these groups with the names of the leading researchers and a short outline of the project.
Applied machine learning in drug discovery. Anthe Janssen (LIC) & Gerard van Westen (LACDR)
In drug discovery and optimization one is often faced with complex high dimensional data. Finding a correct interpretation and a useful extrapolation from this data is almost by definition the strength of machine learning applications. We aim to develop state-of-the-art machine learning tools with a strong focus on making them readily applicable and freely available. Recently, we published one such model (10.1021/acs.jcim.8b00640). We want to continue working on this project and others like it to speed up the practice of drug discovery by providing the bench (bio)chemist with machine learning tools to make informed decisions. Specific challenges in this application are: the noisy nature of biological data (different labs, different assays, etc), the relatively high amount of errors in the data, and the extreme sparseness of the available data.
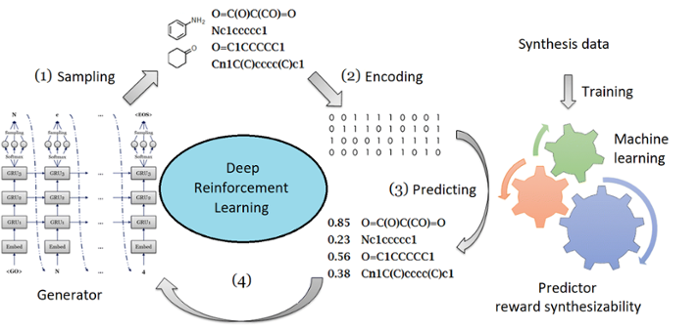
Retrosynthesis and Reinforcement Learning Combined with Mode-of-Action Prediction. Anthe Janssen (LIC), Mike Preuss (LIACS), Mario van der Stelt (LIC) and Gerard van Westen (LACDR)
The ultimate automated drug discovery would be a machine learning application that is capable of generating new molecules and predicting the associated biochemical activities. These new molecules of course need to be synthesizable, soluble and preferably metabolically stable. This results in highly complex multi-objective optimizations. Strategies to solve these optimizations are a big part of Game-AI, which was recently used in one of our retrosynthesis publications (DOI: 10.1038/nature25978). We aim to couple such a retrosynthetic neural network to our recently published DrugEx molecular generator (DOI: 10.1186/s13321-019-0355-6) to be able to prioritize molecules on synthesizability. The DrugEx neural networks can be retrained using reinforcement learning to generate molecules in the chemical space specific for a certain biological target. Ultimately, this setup should get us one step closer to fully automated drug design.