
Research project
Proteochemometrics
Research question
- Contact
- Gerard van Westen
Currently, one of the most widely used approaches to predict the activity of a series of novel chemical compounds on a known protein target is to create a Quantitative Structure Activity Relationship (QSAR) model. This approach, as the name states, relates quantitative properties of a series of ligands to a certain output variable that is considered a measure for activity. However, while this approach has proven to be widely applicable and fast in the past, it has also shown to be somewhat unreliable if used beyond the scope of the original training set.
To circumvent this problem, proteochemometric modeling has been introduced. This approach builds on the same fundamental principles as QSAR, however it adds a quantitative description of the protein in addition to the description of the ligands. The addition of this protein term improves the models and allows for a more reliable extrapolation of the activity of unknown compounds when compared to QSAR on an identical dataset.
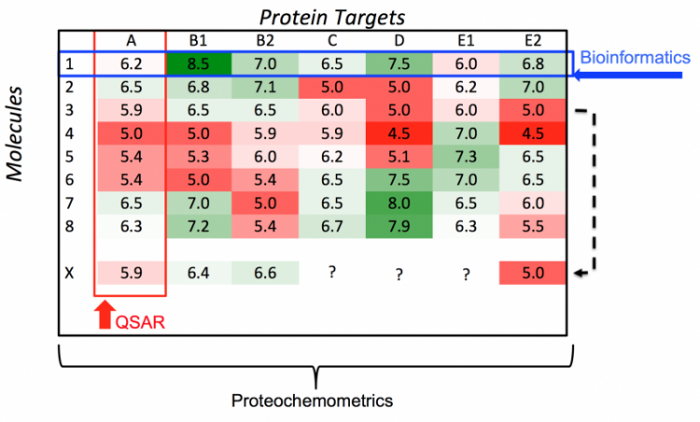
The illustration demonstrates the theory of proteochemometrics and how it builds on the fundamentals of QSAR and bioinformatics. The rows represent chemical compounds 1 – 8 and the columns represent protein targets A – E2. Each field indicates an interaction between a compounds and a protein. The colour of the field indicates whether or not an interaction is favourable / good (green) on unfavourable / bad (red). In a typical QSAR analysis only a single column is considered (several compounds for a single protein). In bioinformatics the interest lies in the rows, i.e. differences / similarities between proteins. Proteochemometrics combines the two and can model the whole matrix, additionally it can be used to make predictions on compound X (indicated by the dotted arrow).
We used proteochemometrics to model several completely different datasets:
- Adenosine receptors (human + rat) & inhibitors
- HIV mutants & preclinical leads
- HIV mutants & clinical drugs
- OATP1B1 / OATP1B3 & inhibitors
References
- van Westen, G.J., Wegner, J.K., IJzerman, A.P., van Vlijmen, H.W. and Bender, A., 2011. Proteochemometric modeling as a tool to design selective compounds and for extrapolating to novel targets. MedChemComm, 2(1), pp.16-30.
- van Westen, G.J., Hendriks, A., Wegner, J.K., IJzerman, A.P., van Vlijmen, H.W. and Bender, A., 2013. Significantly improved HIV inhibitor efficacy prediction employing proteochemometric models generated from antivirogram data. PLoS Comput Biol, 9(2), p.e1002899.