
Research project
Data mining and algorithm development
Due to the modern techniques of combinatorial chemistry and high-throughput screening, data on the biological activity of many millions of compounds is known. However, it is still very difficult to transfer this data into knowledge: if we know that compounds A and B bind to a certain protein with high affinity, and compounds C and D do not interact, and E only slightly, what can we tell about compound F. And, perhaps more importantly, can we use that knowledge to design novel compounds that bind to the chosen receptor with high affinity?
- Contact
- Gerard van Westen
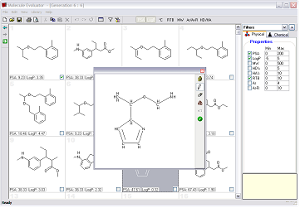
In our group of Medicinal Chemistry we have developed a computer program called the "Molecule Evoluator". Its purpose is to help medicinal chemists design new drugs and ligands that bind to targeted receptors. It does this by mutating and combining structures created by the chemist or the computer program itself and estimating relevant properties of the molecule, like the polar surface area and logP.
We also experiment with several approaches to chemical databases. We have written computer programs for extracting the most frequent chemical substructures from databases like the one of the National Cancer Institute. We may use these substructures in the Molecule Evoluator to generate new molecules that should be more synthetically feasible than those generated from random atoms and substructures. On top of this we added a so-called co-occurrence analysis, and learned which substructures (rings for instance) are frequently found together.
One step further is to allow for more 'abstract' substructures, say "a single or double bond that is connected to a hetero-atom (maybe O, maybe N)". Such 'elaborate graphs' prove to be very useful, as we found out in our first paper that mined for such substructures in a mutagenicity database.