
When semi-local DFT is accurate for dissociative chemisorption on a transition metal surface, and when it is not
Density functional theory (DFT) with so-called semi-local exchange has been remarkable accurate for some dissociative chemisorption reactions on metals, but it has notoriously failed for others. A team of researchers from the University of California at Irvine and the Leiden Institute of Chemistry have discovered for which systems the method is successful, and for which it fails. The team were also able to point to a way forward with treating the "tough-to-handle systems" more accurately in future.
DFT not well understood yet
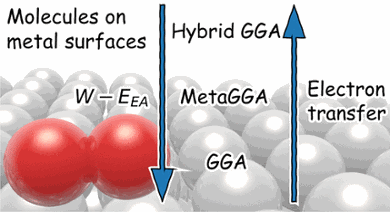
While density functional theory (DFT) is perhaps the most used electronic structure theory in chemistry, many of its practical aspects remain poorly understood. For instance, DFT at the so-called generalized gradient approximation (GGA) tends to fail miserably at describing gas-phase reaction barriers, while it performs surprisingly well for many molecule–metal surface reactions. However, GGA-DFT has also been observed to fail for many systems in the latter category, and up to now it has not been clear when one may expect it to work.
When DFT works for dissociation on metal surfaces
The Leiden-Irvine team have been able to show that GGA-DFT tends to work if the difference between the work function of the metal and the dissociating molecule’s electron affinity is greater than ∼7 eV and to fail if this difference is smaller, with sticking of O2 on Al(111) being a spectacular example that is highlighted in the work. Using dynamics calculations the team show that, for this system, the DFT problem may be solved as done for gas-phase reactions, i.e., by resorting to a more complex and expensive-to-use class of density functionals, i.e., hybrid functionals. To keep computational expense low screening at long-range was used, and this was also done to obtain a correct description of the metal.
Towards an affordable but still accurate approach in future
The team's results further suggest the GGA error in the O2 + Al(111) barrier height to be "functional driven", and not "density driven". The good news is that this suggests the possibility to compute the interaction between the molecule and the surface for the difficult-to-treat systems with computationally cheap so-called non-self-consistent calculations, in which an expensive to use hybrid functional has to be applied only once to a cheap-to-calculate GGA density. This saves at least an order of magnitude in computation time and likely opens the way to a more accurate treatment of the "tough" systems in future.