
Density functional theory is an accurate predictor for variation with geometry of barriers for reactions on metals
A semi-empirical version of the specific reaction parameter approach to density functional theory (SRP-DFT) has been remarkably successful at predicting dissociative chemisorption probability vs. incidence energy curves for reactions on metal surfaces. New quantum Monte Carlo (QMC) calculations on the dissociative chemisorption of H2 on Al(110) show why: With several semi-local density functionals, DFT is remarkably accurate at how the barrier height varies with the geometry of the system.
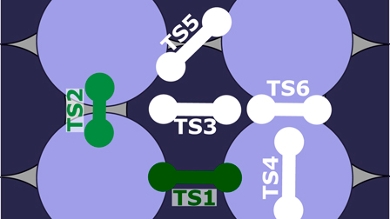
A hard problem: dissociative chemisorption on metals
Reactions of molecules on metal surfaces are notoriously difficult to simulate accurately. Density functional theory can be utilized to generate a potential energy surface, but with presently available functionals using semi-local exchange, the results are not yet accurate enough.
Diffusion Monte-Carlo: a first principles view
To provide benchmark barrier heights for such a problem with a high-quality method, diffusion Monte Carlo (DMC) has been applied to H2 + Al(110). Barrier heights have been computed for six different geometries. The goal of the Leiden University team who carried out the calculations was twofold: first, to provide accurate barrier heights for the two lowest lying transition states of the system investigated, and second, to assess for the chosen system whether density functionals are capable of describing the variation of barrier height with molecular orientation and impact site through a comparison with DMC barriers.
Towards first principle SRP-DFT
Barrier heights computed with selected functionals at the generalized gradient approximation (GGA) and meta-GGA levels have been compared to the DMC results. The comparison shows that all selected functionals yield an accurate description of the variation of barrier height with impact site and orientation. Their absolute values may not be accurate, but this may be remedied by "tuning" the minimum barrier height by choosing an appropriate functional with an adjustable parameter, and adjusting this parameter to fit the QMC barriers as closely as possible. Efforts to fit such a first principles SRP density functional, and to perform dynamics calculations based on this functional and compare with existing experiments on H2 + Al(110), are underway