
Designing better catalysts using simple chemical concepts
An international team of researchers, including Federico Calle-Vallejo of Leiden University’s Institute of Chemistry, have taken the atomic-scale design of catalysts to the next level. Their research contributes to the quest for a method to generate or store energy more efficiently. The report is published in Science on 8 October 2015.
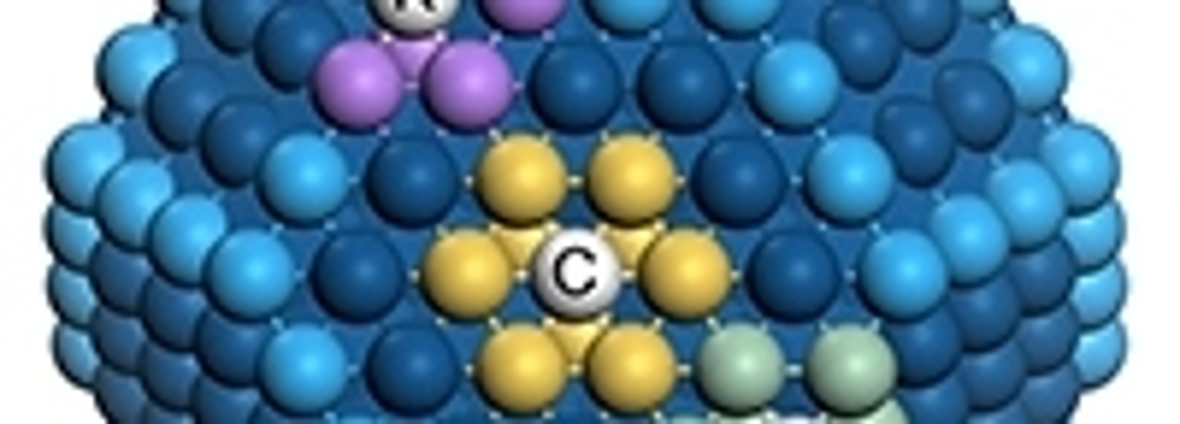
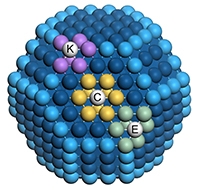 Catalytic nanoparticles contain various sites: for instance, the sites C (centre of a hexagonal facet; nine neighbours are marked in yellow), E (edge between two hexagonal facets; seven neighbours in green) and K (kink, at the corner between three facets; six neighbours in purple). Atoms at edges and terraces are shown in light and dark blue respectively. Each site contributes differently to the total catalytic activity by virtue of its number of neighbours.
Catalytic nanoparticles contain various sites: for instance, the sites C (centre of a hexagonal facet; nine neighbours are marked in yellow), E (edge between two hexagonal facets; seven neighbours in green) and K (kink, at the corner between three facets; six neighbours in purple). Atoms at edges and terraces are shown in light and dark blue respectively. Each site contributes differently to the total catalytic activity by virtue of its number of neighbours.
Why is catalysis important?
Catalysis is essential for modern society. A catalyst can be used to optimise the energy input or output of chemical reactions, and to create valuable new products. This simple idea has inspired people to transform the world.
For instance, the European chemical industry, which is largely based on catalytic processes, generates an annual trade surplus of € 50 billion (1). Apart from the major economic advantages, there is also the benefit of reducing environmental damage: catalysis-based devices, such as exhaust gas converters, help to mitigate the environmental impact of internal combustion engines. In addition, fuel cells are expected to make cars more powerful by simply combining hydrogen and oxygen. These cells are a clean alternative: they can reduce greenhouse gas emissions and bring an end to global dependence on fossil fuels.
Where are we now and where are we heading?
The principle of catalysis currently faces tremendous challenges. Many industrial catalytic processes are not yet optimal, and new, highly active and stable catalysts are required for use in devices such as fuel cells, to make them commercially viable.
The question is therefore: how do we develop new catalysts? They usually consist of small particles with diameters expressed in nanometres or microns. Since the particles’ activity depends on their size, shape and chemical composition (see figure), computational chemistry methodologies are often used to make ‘atom-by-atom’ designs. Ideally, these design routines should find the most active catalysts and indicate the characteristics of optimal sites, so that new, better materials can be created. However, current procedures can only determine the adsorption energetics of optimal catalysts, and not their structure. Developers therefore have to screen large databases to find materials that have these energetics, and then conduct numerous experiments on the most active candidates.
What are the next steps?
Researchers at Leiden University (the Netherlands), Université de Lyon, ENS Lyon, UCB Lyon 1, CNRS (France), Ruhr-Universität Bochum and Technische Universität München (Germany) have taken the atomic-scale design of catalysts to the next level (2). Their design procedure, called “coordination-activity plot”, not only determines the adsorption properties of optimal active sites, but also shows their structure. An additional advantage of this method is that it is based on one of the simplest concepts in chemistry: coordination numbers, which indicate the quantity of atoms in the proximity of the active sites (see examples in the figure).
To test the accuracy of the coordination-activity plots, the researchers computationally designed a new type of platinum catalysts for use in fuel cells. The model catalysts were prepared experimentally using three different synthesis methods, and were found to display high catalytic activity in all cases.
These results herald the start of a new paradigm in catalysis research: the design of materials based on geometric rationales, which give more insight than their energetic equivalents and facilitate the experimental implementation of computational designs.
References
(1) Chlorine Industry Review 2012-2013. Euro Chlor, Belgium, 2013.
(2) Federico Calle-Vallejo, Jakub Tymoczko, Viktor Colic, Quang Huy Vu, Marcus D. Pohl, Karina Morgenstern, David Loffreda, Philippe Sautet, Wolfgang Schuhmann, Aliaksandr S. Bandarenka. Finding optimal surface sites on heterogeneous catalysts by counting nearest neighbors, Science, 2015.