
Research project
Redox catalysis for a sustainable energy infrastructure
The main research theme in the group of Dennis Hetterscheid is to understand and mimic bioinorganic multi-electron processes that are relevant to our future energy infrastructure. Reduction of protons generates hydrogen that can be used as a chemical fuel. Alternatively to gaseous hydrogen, the reduction of carbon dioxide can afford a liquid carbon based fuel. In case of both reactions protons and electrons are necessary which are generated by oxidation of water to produce dioxygen. The group focuses in particular on molecular catalysts that allow for specific structural modifications in order to tune and control the observed catalytic activity.
- Contact
- Dennis Hetterscheid

Bio-inspired and ligand assisted small molecule activation
In nature the role of the protein environment of the catalytic site has several roles. Depending on the electron donating or electron withdrawing character of the ligands coordinated directly to the transition metal active site, the electronic properties of the active site is tuned. Also the geometry of the catalytic site is perfectly oriented for catalysis. These roles of the ligand environment have been well established in coordination chemistry. In addition the protein environment is actively involved in activation of the substrate (e.g. H +, CO 2, N 2, O 2 or H 2O) via hydrogen bonding networks, and shuttling protons from or to the active site. The latter events are critical for efficient multi-electron catalytic processes, yet have hardly been explored. In my group molecular catalysts are studied that are employed with ligands that bear functionalities in the second coordination sphere. These functionalities are positioned as such that they assist in activation of the substrate and can shuttle protons away from the catalytic site in case of oxidation reactions and to the active site in case of the reduction reactions.
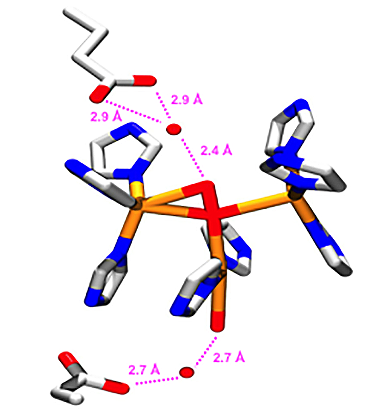
Enzyme mimics
Since natural multi-electron redox catalysts operate very efficiently, such systems form a major inspiration for development of new catalytic systems. In addition fundamental studies on these enzyme mimics will indirectly reveal how catalysis in nature may occur. Currently, my group is interested in model systems of the oxygen evolving center of photosystem II, and the active sites of the dioxygen reduction enzymes Laccase and cytochrome c oxidase.
Electrochemistry and in situ spectroscopy
To catalyze redox half reactions we rely on electrochemical techniques to study the rates of the catalytic reaction as a function of applied potential and the number of active sites involved. Our focus lies with fundamental understanding of these systems and with unraveling the mechanisms wherein these bio-inspired molecular redox catalysts operate. To determine the structure of the catalytic species that are present in the reaction mixture at a particular applied potential we use Raman spectroscopy, IR spectroscopy and UV spectroscopy coupled to electrochemistry. In combination with reaction kinetics by means of various electrochemical techniques gives us a clear picture how catalysis proceeds. Typically several catalytic intermediates are transient species and cannot be observed experimentally. To shed light on these intermediate species of the catalytic cycle we rely on density functional theory. In our approach we use experimental data to calibrate our computational model.
Synthesis and structure activity studies
A major advantage of molecular catalysts is that these can be varied to a large degree in order to radically improve the catalytic activity of the targeted multi-electron redox reaction. For example implementation of a proton delivery or removal system may seriously affect the reaction rates. An important research question is how and to which extend a particular ligand modification affect the rate determining step, or bottleneck, of the catalytic reaction. Another important parameter is the potential that is required to achieve a particular catalytic rate. We aim to conduct the chemical reaction of choice at the mildest conditions possible, as this will result in the highest efficiency in terms of energy consumption. This latter is the major crux in the formation of chemical fuels from renewable energy and the subsequent consumption of these chemical fuels.
Key publications
-
Venturini, A., A. Barbieri, J.N.H. Reek, D.G.H. Hetterscheid, "Catalytic Water Splitting with an Iridium Carbene Complex: A Theoretical Study", Chemistry - A European Journal, vol. 20, issue 18, pp. 5358 - 5368, 04/2014. DOI: 10.1002/chem.201303796
-
Hetterscheid, D.G.H., J.N.H. Reek, "Periodate as an Oxidant for Catalytic Water Oxidation: Oxidation via Electron Transfer or O-Atom Transfer?", European Journal of Inorganic Chemistry, vol. 2014, issue 4, pp. 742 - 749, 02/2014, 2013. DOI: 10.1002/ejic.201300249
-
Hetterscheid, D.G.H., J.N.H. Reek, "Mononuclear Water Oxidation Catalysts", Angewandte Chemie-International Edition, vol. 51, no. 39, pp. 9740-9747, 2012. DOI: 10.1002/Anie.201202948
-
Hetterscheid, D.G.H., J.N.H. Reek, "Me-2-NHC based robust Ir catalyst for efficient water oxidation", Chemical Communications, vol. 47, no. 9, pp. 2712-2714, 2011. DOI: 10.1039/C0cc05108j